
Introduction: What Corrective Action Means in Manufacturing
A defect may be reworked in one shift, but if the same issue shows up again next week, the factory has not really solved anything. That is why corrective action matters in manufacturing: it is not just about fixing a bad part, stopping a line, or closing a complaint quickly. It is about identifying why the problem happened and removing the cause so it does not keep returning through scrap, rework, customer complaints, audit findings, or NCRs.
In practical terms, corrective action starts after a nonconformance is found and asks a deeper question than immediate containment. Replacing a faulty component, sorting suspect stock, or issuing a one-time operator reminder may address the symptom. A proper quality corrective action goes further by tracing the issue to its root cause, whether that is an unclear work instruction, tool wear, supplier variation, or an uncontrolled process change.
This matters because repeat quality issues are expensive. Poor quality is estimated to cost manufacturers 15% 至 20% 的销售收入 in many operations when scrap, rework, downtime, returns, and warranty costs are added together. In the sections that follow, we will clarify how corrective action differs from correction and preventive action, when the process should begin, and how quality teams can track it from investigation through closure.
Correction, Corrective Action, and Preventive Action: What’s the Difference?
In manufacturing, these three terms are related but not interchangeable. A correction deals with the immediate nonconformance, a corrective action removes the cause of a problem that already happened, and a preventive action reduces the chance of a similar problem happening elsewhere or in the future. If your team mixes them up, the corrective action process often stops at containment and never reaches lasting process improvement.
A Simple Comparison Framework
A practical way to separate the three is to ask three questions. What do we need to fix right now? That is the correction. Why did this happen, and what must change so it does not recur? That is the corrective action. Where else could the same failure mode appear, and how do we stop it before it spreads? That is preventive action within a broader corrective and preventive action system.
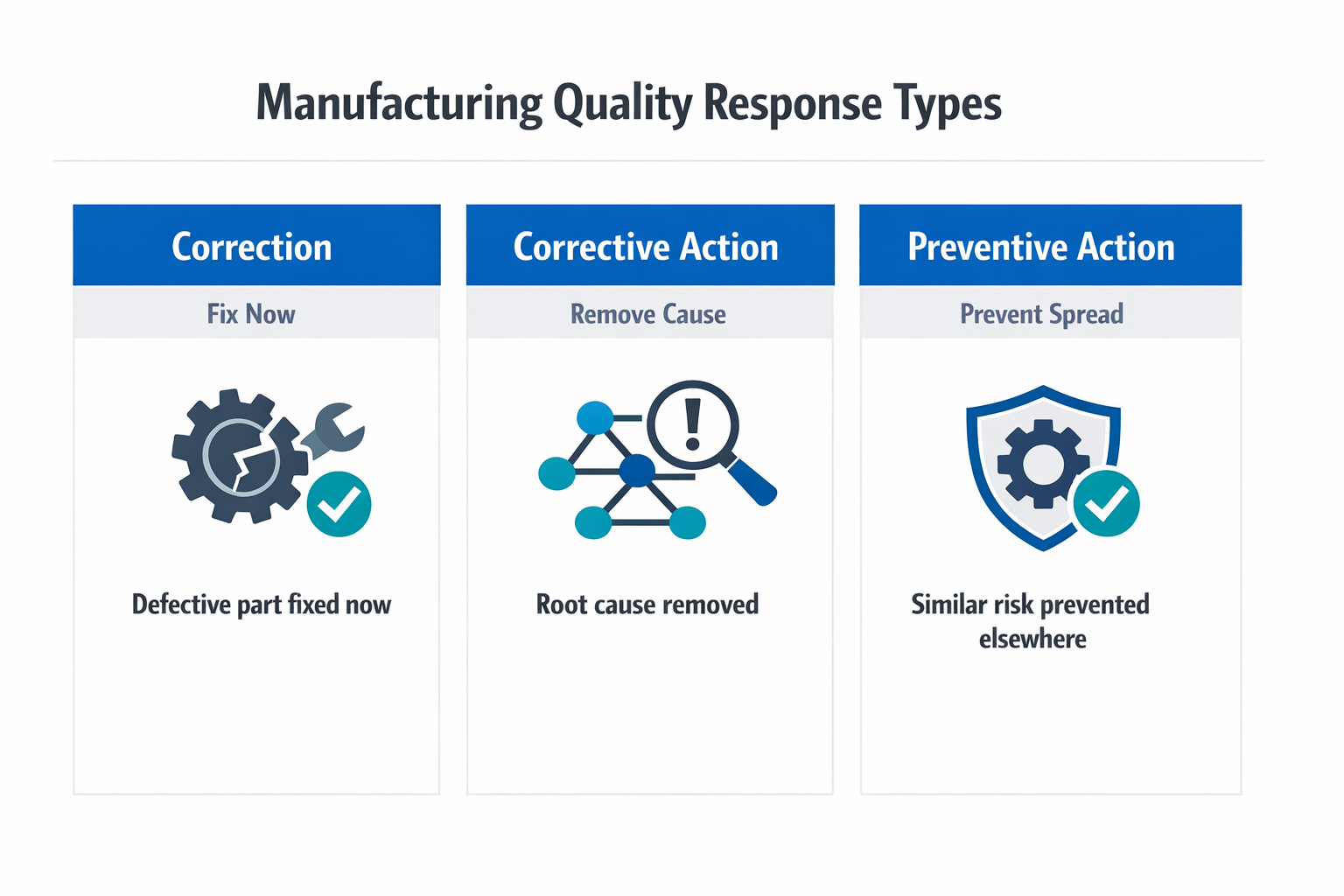
This distinction matters because many quality teams close issues too early. Replacing defective parts, sorting stock, or reworking output may restore shipment status, but those steps do not by themselves count as quality corrective action. The real shift happens when root cause analysis turns an isolated response into a controlled change in method, training, tooling, inspection, or supplier management.
Customer Complaint Example
A customer reports that finished housings arrived with scratched surfaces. The correction is to sort affected inventory, replace damaged units, and protect any open shipments. The corrective action might be changing the packaging method after root cause analysis shows the scratches came from loose part movement during transit, not from machining. A preventive action could then extend the revised packaging standard to similar product families that use the same tray design.
Supplier Defect Example
A batch of incoming fasteners fails hardness inspection. The correction is to quarantine the lot and block it from production use. The corrective action may involve updating the supplier control plan, tightening incoming inspection rules, or requiring process capability evidence if the root cause analysis shows unstable heat treatment at the supplier site. The preventive action would be reviewing whether other critical fastener suppliers carry the same risk and applying the same control logic before another escape occurs.
Internal Process Deviation Example
On an electronics line, operators find solder bridges above the normal defect threshold during in-process inspection. The correction is immediate rework and temporary containment of boards produced in that time window. The corrective action could be revising stencil cleaning frequency and oven parameter checks after investigation confirms process drift, while the preventive action may include updating the control plan and training matrix across all similar SMT lines. That is where corrective action tracking becomes important: teams need to confirm not only that the fix was implemented, but that the same defect rate does not return.
Where Root Cause Analysis Fits
Root cause analysis is the bridge between short-term response and long-term control. Methods such as the 5 Whys 或 fishbone diagram help teams avoid shallow conclusions like “operator error” and identify the system condition behind the failure. In practice, strong corrective action examples usually show a clear chain from nonconformance to cause, then from cause to verified process change, while weak ones stop at rework or retraining without evidence that the issue is truly resolved.
When the Corrective Action Process Should Start
Triggers That Require Formal Corrective Action
A formal corrective action process should start when the issue signals a system weakness, not just a one-time miss. In manufacturing, the most common triggers are recurring defects, nonconformance reports (NCRs), audit findings, customer complaints, supplier escapes, and process deviations that affect product quality, compliance, delivery, or safety. If the same problem appears across multiple batches, shifts, lines, or suppliers, that is usually a clear sign that simple rework or containment is no longer enough. This is where quality corrective action moves from fixing output to addressing the underlying process failure.
Recurring internal defects are often the earliest warning sign. For example, if an injection molding line keeps producing short shots on the same cavity over three shifts, the first response may be segregation and machine adjustment, but repeat occurrences should trigger a formal investigation. The same applies when an NCR shows repeated dimensional failure on a machined part or when SPC data shows a drift that operators keep correcting without stabilizing the process. In these cases, root cause analysis should begin before the issue becomes a customer-facing problem.
External triggers usually require faster escalation because the cost of recurrence is higher. A customer complaint about wrong torque on a fastener, a supplier lot with mislabeled material, or an audit finding tied to missing traceability records can all justify immediate entry into a corrective and preventive action workflow. In regulated sectors such as medical devices, food, or aerospace, even a single documented deviation may require formal action because of compliance exposure. The threshold is lower when the issue affects safety, traceability, or regulatory obligations.
When a Simple Correction Is Enough
Not every problem needs a full corrective action record. If the issue is isolated, low risk, easily contained, and unlikely to recur, a simple correction may be the right response. For instance, replacing one damaged label during final packing or reprinting a traveler with no impact on traceability may be closed at the supervisor level if there is no broader pattern. The key question is whether the event points to a broken control, unclear standard, or unstable process.
Teams should escalate when one or more of these conditions exist: repeat occurrence, customer impact, cross-functional involvement, unclear cause, compliance risk, or significant scrap, downtime, or rework cost. A practical rule many plants use is to formalize any issue that crosses a severity threshold or repeats more than once within a defined period. That keeps the system focused on meaningful risks instead of turning every minor defect into administrative work. It also makes corrective action tracking more useful because the database reflects real systemic issues.
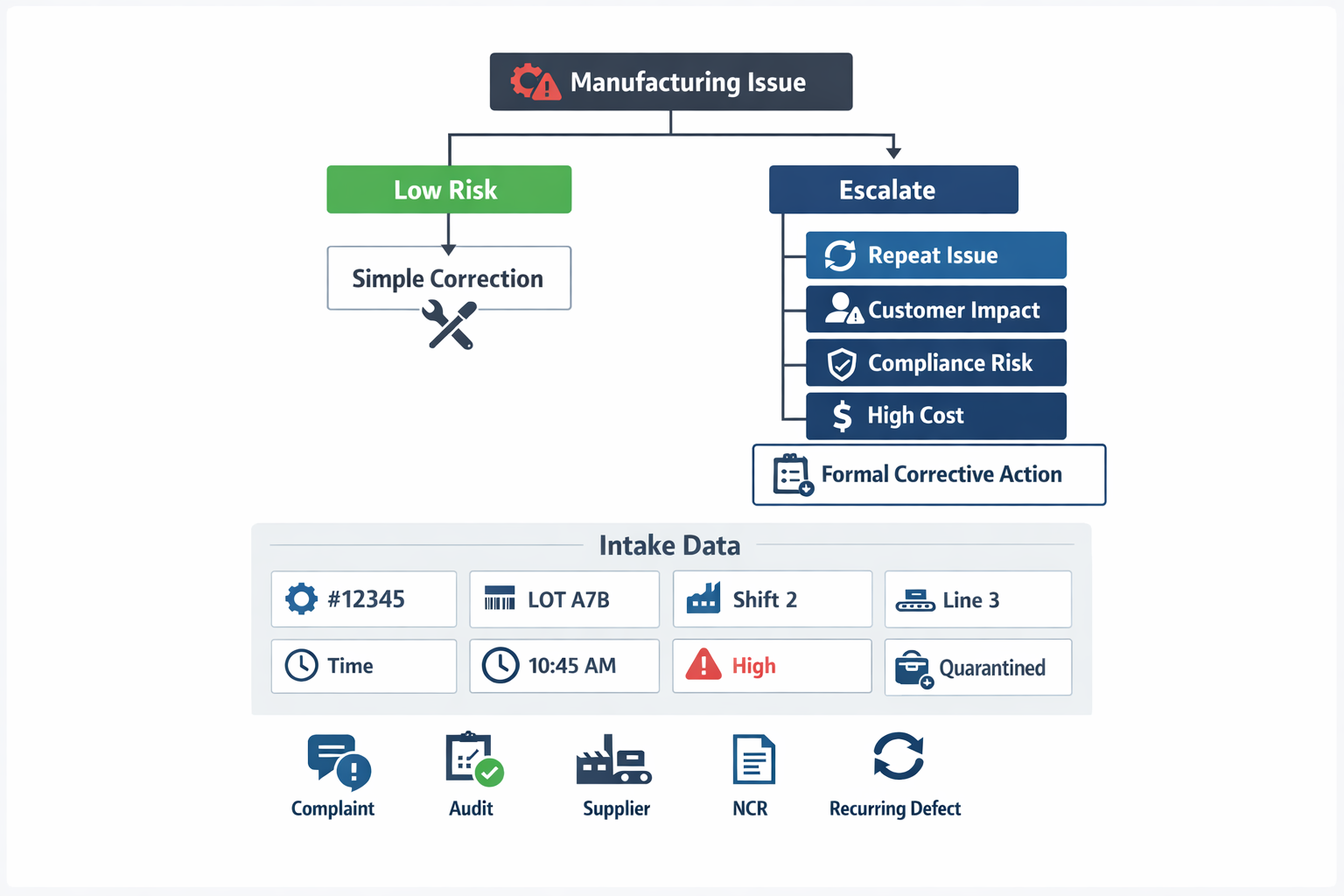
What Quality Teams Should Capture at Intake
The intake step should capture enough information to support fast containment and later investigation. At minimum, teams should log the part number, batch or lot number, shift, production line or machine, date and time detected, issue description, severity, immediate containment action, and the responsible owner. If the issue involves supplier material, customer returns, or audit findings, the source should also be recorded at intake so the case can be routed correctly. Missing these basics is one of the main reasons corrective action stalls before root cause analysis even starts.
Good intake data also helps teams decide whether the case belongs in routine nonconformance handling or in a formal corrective action process. For example, if a stamping defect is tied to one coil from one supplier lot, supplier quality may need to lead the case. If the same defect appears across two presses and multiple operators, manufacturing engineering may need ownership instead. Clear intake fields reduce handoff delays and make later corrective action examples easier to compare across plants, lines, and suppliers.
A Step-by-Step Corrective Action Process for Quality Teams
强大的 corrective action process follows a clear sequence: identify the issue, contain the risk, investigate the cause, assign actions, implement changes, verify results, and formally close the case. In practice, this sequence keeps quality corrective action from stopping at paperwork or rework and pushes the team toward a repeatable fix. To make that concrete, follow one example through the full process: a machining line producing aluminum housings with an out-of-tolerance bore diameter that has triggered repeated internal rejects and one customer return.
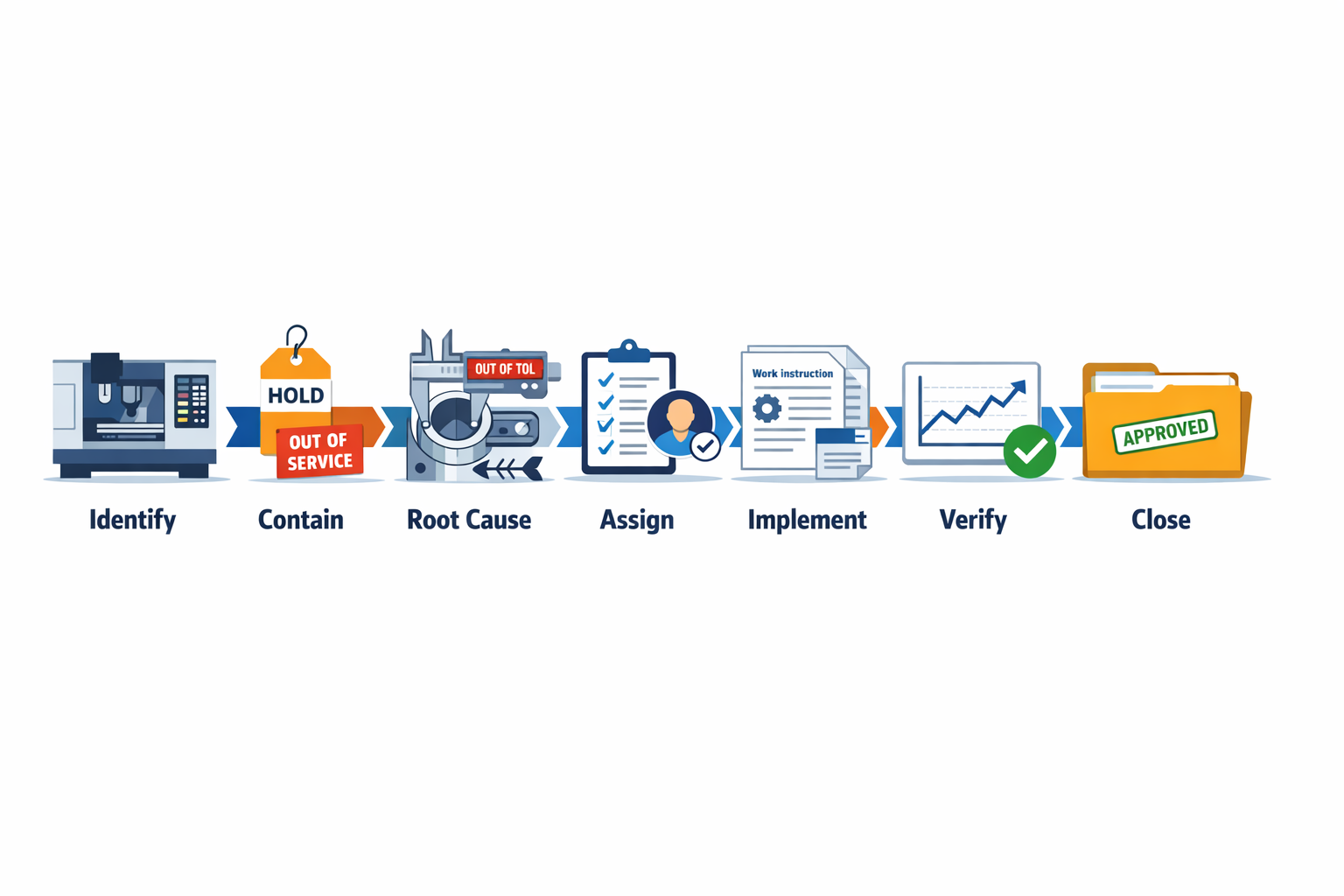
Identify the Issue and Contain the Risk
The first step is to document the nonconformance with enough detail to support action, not just record that “parts are bad.” For the housing defect, the quality engineer logs the part number, machine ID, batch numbers, inspection results, defect rate, affected shift, and whether suspect stock has already moved to assembly or shipment. Good intake data reduces delays later, especially when multiple departments need to respond quickly.
Containment comes next because the plant must stop the problem from spreading while the team investigates. In this example, the supervisor places in-process and finished stock on hold, starts a 100% inspection on the last three lots, and notifies planning that output from the affected CNC line may be constrained. Containment is not the corrective action itself; it is a temporary control that protects the customer and buys time for proper investigation.
Investigate the Root Cause
Once the line is stable, the team moves into root cause analysis. A practical approach is to start with the 5 Whys for speed, then use a fishbone diagram if the issue may involve multiple variables such as machine condition, tooling, method, measurement, material, or operator practice. The goal is to identify the process failure that allowed the defect to occur and escape, not just the point where it was detected.
In the housing example, the team asks why the bore diameter drifted beyond tolerance. The investigation shows the cutting tool wore faster than expected, but that alone is not the full cause; the deeper issue is that the tool-change frequency in the work instruction was based on an older material lot and had not been revised after a supplier material hardness change. A second escape point also appears: first-piece verification was completed, but no in-process check was required after every set number of cycles, so the drift was not caught early.
Assign Owners and Define Corrective Actions
After the causes are confirmed, each action needs a clear owner, due date, and expected outcome. Without that structure, corrective and preventive action efforts often become a discussion rather than an execution plan. Quality should coordinate the record, but actions usually sit across production, engineering, maintenance, supplier quality, and training.
For this case, manufacturing engineering owns the update to tool life standards and work instructions, production owns the revised in-process inspection frequency, procurement and supplier quality review the incoming material change controls, and training leads operator requalification on the updated checks. Each action should state what will change in the process, what evidence will prove completion, and what risk is being addressed. This is where corrective action tracking becomes essential, because accountability depends on visible status rather than verbal follow-up.
Implement the Changes in the Process
Implementation is the point where many corrective actions weaken, because teams close tasks based on intent instead of confirmed execution. The plant should verify that documents are revised, tooling parameters are changed at the machine, inspection plans are updated in the quality system, and affected employees have actually been trained. If one action depends on another, the sequence should be controlled so the line does not restart under mixed conditions.
In the machining example, the team updates the standard tool replacement interval from 1,200 cycles to 800 cycles for the harder material range, adds a mandatory bore check every 200 cycles, and installs a visual counter prompt at the machine. The old work instruction is withdrawn, the quality plan is updated, and the supervisor signs off that all three operators on the line completed retraining before normal production resumes. These details matter because a corrective action process only works when the process on the shop floor is different after the action than it was before.
Verify Effectiveness Before Closure
Implementation does not prove the corrective action worked; verification does. Quality teams should define an effectiveness check up front, usually tied to time, volume, or recurrence criteria, such as zero repeats across the next five lots or stable process capability over two weeks. This step separates a completed task from an effective corrective action.
For the housing line, quality monitors the next eight production lots and reviews bore-diameter measurements, scrap rate, and customer returns. The line shows no repeat nonconformance, in-process checks are completed on time, and process capability improves from a marginal level to an acceptable range. If the defect reappears, the case should be reopened or escalated rather than closed as “done.”
Close the Record and Capture Learning
Formal closure should happen only after evidence is complete: containment record, investigation summary, root cause analysis, implemented actions, effectiveness results, and management approval where required. This creates an audit-ready history and helps future teams understand what was changed and why. It also supports trend review across similar issues, which is one reason corrective action tracking matters beyond the single incident.
In this example, the case is closed only after the quality manager confirms that the revised control plan, updated work instruction, operator training records, and verification data are attached to the record. The final lesson is also shared with another machining cell using the same material family, reducing the chance of a parallel failure elsewhere. That is the difference between isolated firefighting and a disciplined corrective action process.
Corrective Action Examples, Common Breakdowns, and the Metrics That Matter
Corrective Action Examples Across Different Manufacturing Environments
In automotive assembly, a recurring door misalignment issue is a good test of whether a corrective action process is actually working. Weak follow-up would stop at rework, operator retraining, and a note in the shift report. Strong quality corrective action goes further: the team confirms the torque tool drifted out of spec, updates the calibration interval, adds an error-proofing check at the station, and verifies defect rates over the next production runs. The difference is simple—one response fixes the symptom, while the other removes the condition that keeps recreating the defect.
In electronics manufacturing, solder voids on PCB assemblies often trigger repeated customer returns if the response stays too narrow. A poor response might blame operator technique and ask for more inspection, even when the real issue is unstable reflow oven temperature or an unsuitable solder paste storage practice. A stronger response combines root cause analysis with process validation: check thermal profiles, review material handling records, revise setup parameters, and confirm that first-pass yield improves after implementation. This is where corrective and preventive action start to overlap in practice, because a robust fix often strengthens controls beyond the single incident.
In medical device manufacturing, documentation gaps can be just as serious as physical defects because they create compliance risk and product traceability issues. If a device history record is incomplete, weak follow-up may end with a late signature and a reminder email to staff. Strong corrective action examines why the record was missed in the first place, such as unclear approval rules, poor form design, or uncontrolled handoffs between production and QA. The action is only complete when the revised workflow consistently prevents missing fields or unauthorized release.
Where Manual Corrective Action Systems Break Down
The most common breakdown in manual systems is not the lack of effort; it is the lack of control. Many factories still manage corrective action tracking through email threads, shared spreadsheets, printed NCR forms, and meeting notes, which makes it hard to see status across departments. Once multiple owners, due dates, and verification steps are involved, version confusion becomes almost inevitable. In practice, that means actions stay “open” long after the issue appears closed on the shop floor.
Ownership is another frequent failure point. When the corrective action process does not clearly separate the investigator, action owner, approver, and verifier, tasks get pushed sideways between quality, production, maintenance, and supplier quality teams. That creates delays, especially when one action depends on another, such as maintenance completing a machine fix before QA can validate effectiveness. The result is overdue actions that are not visible until the next audit, customer complaint, or management review.
Verification is often the weakest link. Teams may document that an action was completed, but they do not always confirm whether it actually reduced recurrence, scrap, complaints, or deviation rates. ISO-oriented quality systems expect evidence of effectiveness, not just evidence that a task was assigned and marked done. Without that check, the same nonconformance can return under a different batch, shift, or customer order.
The KPIs That Show Whether Corrective Action Is Working
A small set of operational KPIs can show whether your corrective action system is improving process control or just generating paperwork. Closure time measures how long issues remain open from initiation to approved closure, while overdue action rate shows whether assigned tasks are being completed on time. Repeat issue rate is especially important because repeated defects usually signal weak root cause analysis or poor implementation discipline. Effectiveness check pass rate helps quality leaders see whether closed actions are actually holding after a defined monitoring period.
These metrics are more useful when segmented by plant, line, defect type, supplier, or department. For example, a factory may show acceptable average closure time overall but still have chronic delays in supplier-related actions or engineering changes. According to industry quality management benchmarks, delayed closure and repeated nonconformances are among the clearest indicators of an immature CAPA discipline because they reflect both process speed and process quality. In other words, fast closure is not enough if the same issue comes back.
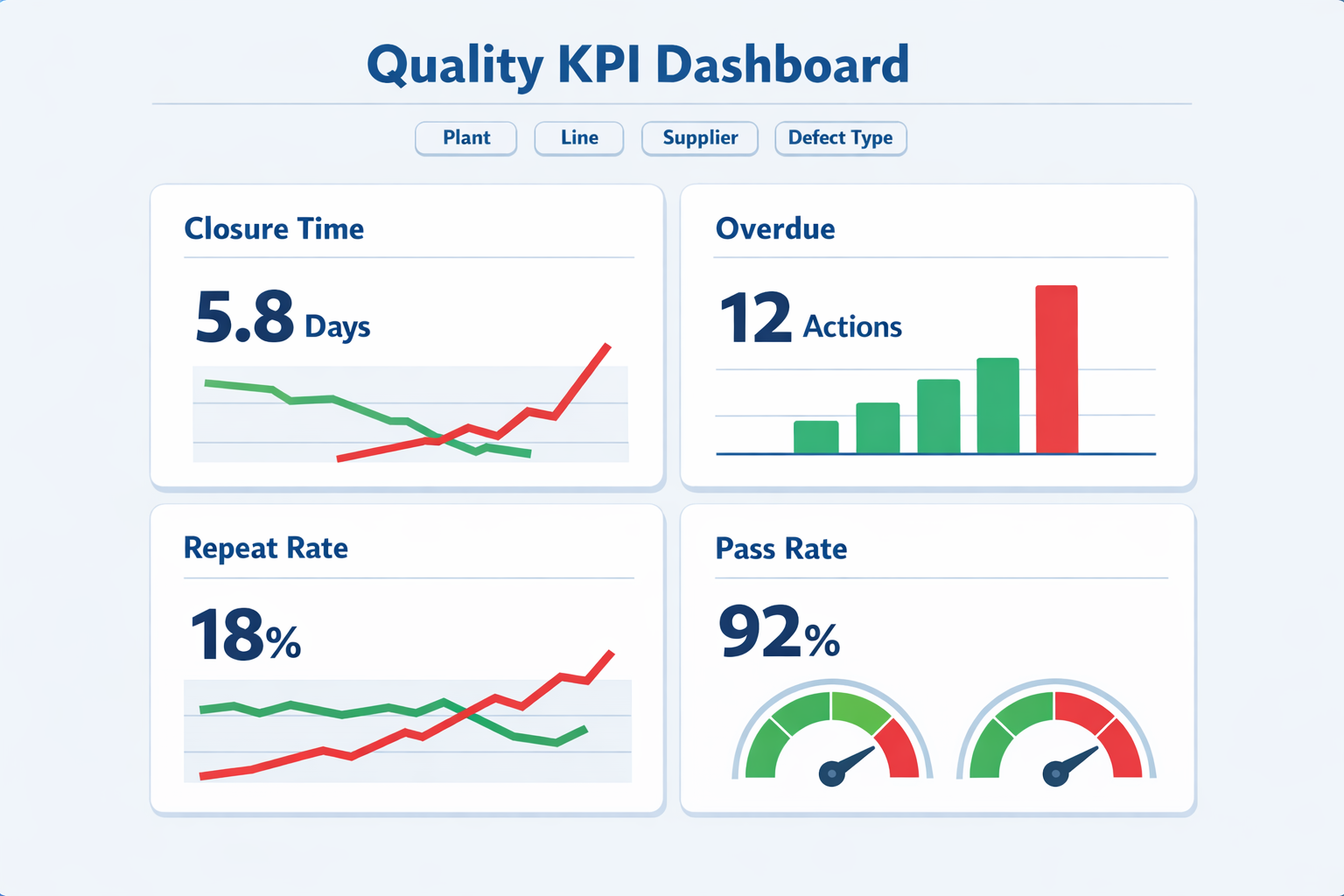
For management review, the best reporting view is usually a mix of volume, timeliness, and effectiveness. That means tracking how many corrective actions were opened, how many remain overdue, what percentage passed verification, and how many similar issues recurred within 30, 60, or 90 days. When those four signals are reviewed together, plant leaders can quickly tell whether the system is reducing risk or simply moving forms through approval steps.
Conclusion: How Jodoo Helps Manufacturers Digitize Corrective Action Tracking
强大的 corrective action process needs more than a form, an email trail, and a spreadsheet owner list. To prevent repeat defects, quality teams need one connected system for issue intake, containment, root cause analysis, action assignment, verification, and closure. That is what turns corrective action from reactive firefighting into a controlled quality process that stands up during audits and management reviews.
Jodoo helps manufacturers digitize corrective action tracking without custom development. Using no-code forms, you can capture NCRs, complaints, audit findings, and supplier issues with the right fields from the start, then route each case through investigation, approvals, due dates, reminders, and effectiveness checks. Managers can also use dashboards to track closure time, overdue actions, repeat issues, and open cases by plant, line, supplier, or severity.
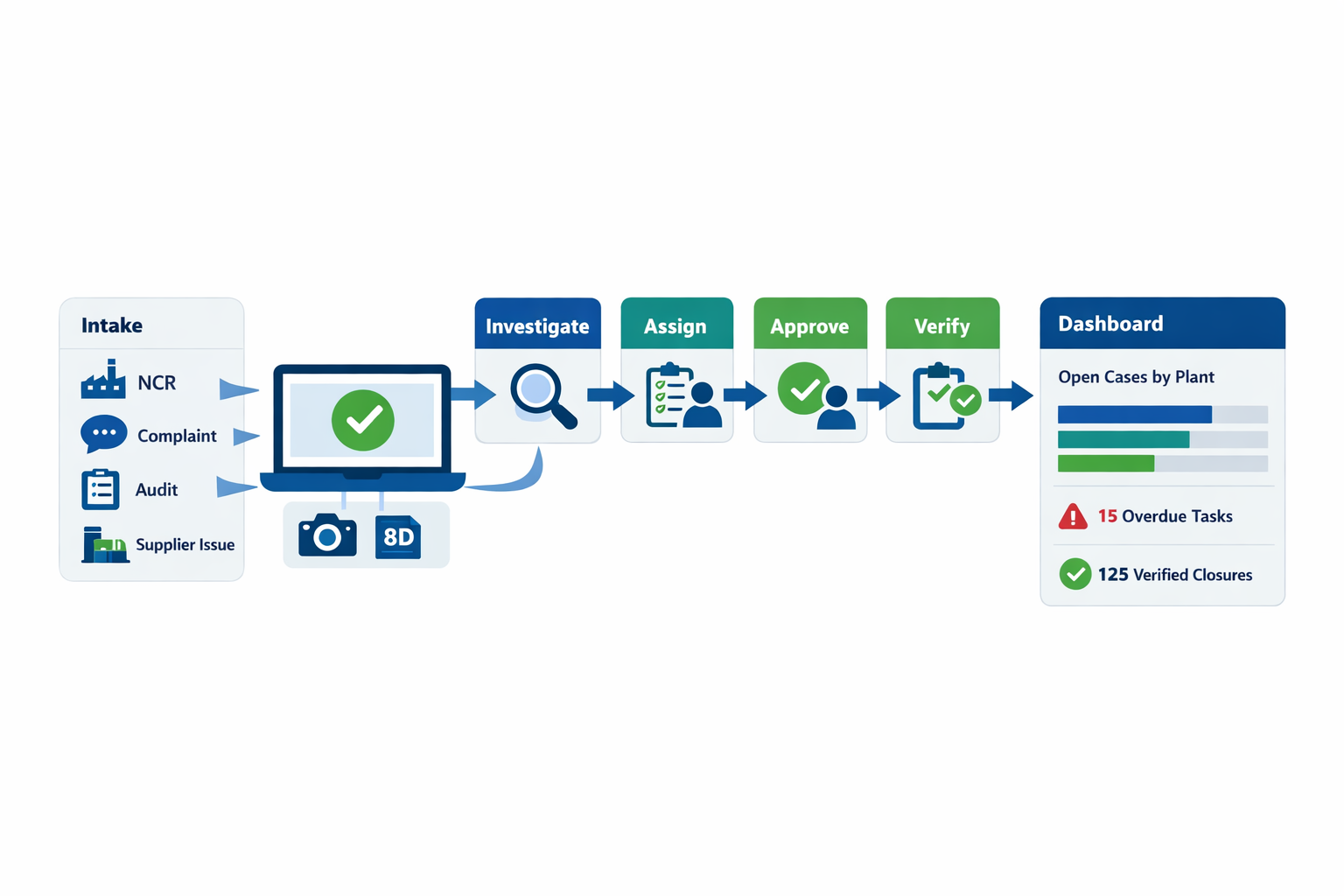
For example, a supplier quality team can use Jodoo to log a complaint from incoming inspection, trigger an investigation workflow, assign tasks to SQE and production, attach photos and 8D records, and verify closure before the case is marked complete. The result is faster follow-up, clearer accountability, and audit-ready records in one place.
If you want a practical way to standardize corrective action across sites, Jodoo is a flexible no-code lean manufacturing platform worth exploring. 开始免费试用 或者 预约演示 now.
